ESgamess: molecule and Gamess job handling class (basic demo)
30/03/21
This class handles Gamess jobs (full pipeline) on a local machine only.
Core functionality is provided by the following libraries:
-
Interface with PubChem.
-
Molecule class/handling routines.
Transformations.
Figures (2D natively, uses py3Dmol on the backend for 3D rendering).
-
Setup Gamess input cards.
Run Gamess calculations (local machine only).
This class creates a pipeline with these tools, and implements a few extra helper routines, with the general aim to make this part of the process as painless as possible.
Minimal method for pipeline to ePolyScat jobs:
PubChem download > Fix reference frame (symmetry axis to Z) > Run Gamess > Export/convert.
This is implemented directly.
22/11/23, 23/01/24 (debug & tidy)
Updated some methods and finished notes.
Better setCoords() handling, including Pandas method.
Support for XYZ files for molecule creation (RDkit version > 2022.03 required).
Imports
[1]:
# Import class
from epsman.elecStructure.gamess import ESgamess
Molecule creation routines
Currently wraps routines from RDkit + PubChemPy for rapid setup from existing sources. Shows 2D structure and coord tabe on execution.
Notes:
Atom sequence and labelling may change with method.
Similarly, atomic charges, bonding and display may change with method.
In general, this doesn’t matter if coords are passed to another electronic structure routine, but may be important in some cases.
TODO: add support for manual molecule creation. This can be done via a file at the moment, or via RDkit backend (see, for example, the RDkit docs).
Easy methods
[2]:
# Molecule from PubChem
testDL = ESgamess(searchName = 'N2O')
Set name = N2O
Set smiles = None
Set molFile = None
Set pd = None
Set xyz = None
Set molOverride = None
*** File /mnt/femtobackSSHFS/DriveSyncShare/code-share/github-share/epsman/docs/doc-source/demos/N2O.SDF already exists; existing file will be used. Pass overwrite=True to overwrite.
Set job = None
Set sym = C1
Set atomList = None
Set precision = 6
Set atomsDict = {}
Set refDict = {}
Set atomsHist = {}
*** Updating coords (Pandas version).
[14:15:07] Warning: molecule is tagged as 3D, but all Z coords are zero
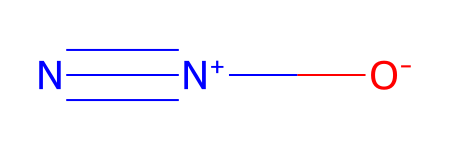
| Ind | Species | Atomic Num. | x | y | z | |
|---|---|---|---|---|---|---|
| 0 | 0 | O | 8 | 1.3063 | 0.0 | 0.0 |
| 1 | 1 | N | 7 | -0.1096 | 0.0 | 0.0 |
| 2 | 2 | N | 7 | -1.1967 | 0.0 | 0.0 |
[3]:
# Molecule from SMILES
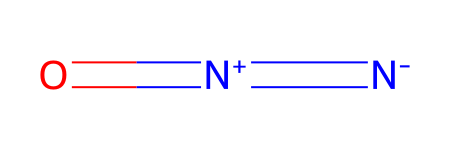
testSmiles = ESgamess(smiles = '[N-]=[N+]=O')
Set name = None
Set smiles = [N-]=[N+]=O
Set molFile = None
Set pd = None
Set xyz = None
Set molOverride = None
Set job = None
Set sym = C1
Set atomList = None
Set precision = 6
Set atomsDict = {}
Set refDict = {}
Set atomsHist = {}
*** Updating coords (Pandas version).
| Ind | Species | Atomic Num. | x | y | z | |
|---|---|---|---|---|---|---|
| 0 | 0 | N | 7 | 1.222602 | 0.0 | 0.0 |
| 1 | 1 | N | 7 | -0.007294 | 0.0 | 0.0 |
| 2 | 2 | O | 8 | -1.215308 | 0.0 | 0.0 |
[4]:
# From file, e.g. SDF file downloaded from PubChem above.
# This uses RDkit Chem.MolFromMolFile() on the backend,
# For details of files supported see https://www.rdkit.org/docs/RDKit_Book.html#mol-sdf-support-and-extensions
# and https://www.rdkit.org/docs/source/rdkit.Chem.rdmolfiles.html
testFile = ESgamess(molFile = 'N2O.SDF')
Set name = None
Set smiles = None
Set molFile = N2O.SDF
Set pd = None
Set xyz = None
Set molOverride = None
Set job = None
Set sym = C1
Set atomList = None
Set precision = 6
Set atomsDict = {}
Set refDict = {}
Set atomsHist = {}
*** Updating coords (Pandas version).
[14:15:07] Warning: molecule is tagged as 3D, but all Z coords are zero
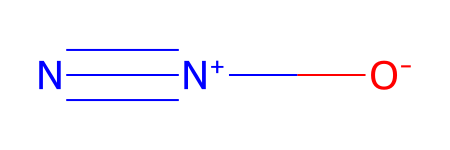
| Ind | Species | Atomic Num. | x | y | z | |
|---|---|---|---|---|---|---|
| 0 | 0 | O | 8 | 1.3063 | 0.0 | 0.0 |
| 1 | 1 | N | 7 | -0.1096 | 0.0 | 0.0 |
| 2 | 2 | N | 7 | -1.1967 | 0.0 | 0.0 |
Manual or from XYZ file
Since RDKit 2022.03 XYZ file support is included, which is also suitable for manual molecule creation from a list of atoms and coordinates.
Full details can be found in https://github.com/rdkit/UGM_2022/blob/main/Notebooks/Landrum_WhatsNew.ipynb
The method can be used to init a system from a file or string representation. The basic format is:
[no. atoms]
[Atom 0] [x] [y] [z]
[Atom 1] [x] [y] [z]
...
[5]:
# Generate XYZ representation from existing case above
testDL.genXYZ()
Generated XYZ string repr:
3
O 1.3063 0.0 0.0
N -0.1096 0.0 0.0
N -1.1967 0.0 0.0
Data set to self.xyzStr
[6]:
# Use the string representation to define a new system
testXYZstr = ESgamess(xyz = testDL.xyzStr)
Set name = None
Set smiles = None
Set molFile = None
Set pd = None
Set xyz from passed values.
Set molOverride = None
Set job = None
Set sym = C1
Set atomList = None
Set precision = 6
Set atomsDict = {}
Set refDict = {}
Set atomsHist = {}
*** Updating coords (Pandas version).
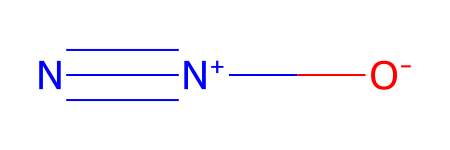
| Ind | Species | Atomic Num. | x | y | z | |
|---|---|---|---|---|---|---|
| 0 | 0 | O | 8 | 1.3063 | 0.0 | 0.0 |
| 1 | 1 | N | 7 | -0.1096 | 0.0 | 0.0 |
| 2 | 2 | N | 7 | -1.1967 | 0.0 | 0.0 |
[7]:
# Note that this routine can accept a string, or filename.
testXYZfile = ESgamess(xyz = 'N2O.xyz')
Set name = None
Set smiles = None
Set molFile = None
Set pd = None
Set xyz from passed values.
Set molOverride = None
Set job = None
Set sym = C1
Set atomList = None
Set precision = 6
Set atomsDict = {}
Set refDict = {}
Set atomsHist = {}
*** Updating coords (Pandas version).
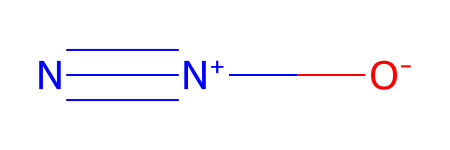
| Ind | Species | Atomic Num. | x | y | z | |
|---|---|---|---|---|---|---|
| 0 | 0 | O | 8 | 1.3063 | 0.0 | 0.0 |
| 1 | 1 | N | 7 | -0.1096 | 0.0 | 0.0 |
| 2 | 2 | N | 7 | -1.1967 | 0.0 | 0.0 |
[8]:
# Creation via a Pandas representation is also available
# (as of 25/01/24, https://github.com/phockett/epsman/commit/bd21149f2aa6ee4ea23ab4dfd3f4f4a60503508e)
# Pandas table format as shown above, and in self.pdTable
testPDmol = ESgamess(pd = testXYZfile.pdTable)
Set name = None
Set smiles = None
Set molFile = None
Set pd from passed values.
Set xyz = None
Set molOverride = None
Set job = None
Set sym = C1
Set atomList = None
Set precision = 6
Set atomsDict = {}
Set refDict = {}
Set atomsHist = {}
*** Updating coords (Pandas version).
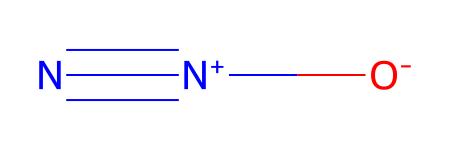
| Ind | Species | Atomic Num. | x | y | z | |
|---|---|---|---|---|---|---|
| 0 | 0 | O | 8 | 1.3063 | 0.0 | 0.0 |
| 1 | 1 | N | 7 | -0.1096 | 0.0 | 0.0 |
| 2 | 2 | N | 7 | -1.1967 | 0.0 | 0.0 |
Additional info
If Pandas is available, a fancy print is available with printTable().
[9]:
testDL.printTable()
| Ind | Species | Atomic Num. | x | y | z | |
|---|---|---|---|---|---|---|
| 0 | 0 | O | 8 | 1.3063 | 0.0 | 0.0 |
| 1 | 1 | N | 7 | -0.1096 | 0.0 | 0.0 |
| 2 | 2 | N | 7 | -1.1967 | 0.0 | 0.0 |
If py3Dmol is available, interactive 3D plots are available in notebooks with plot3D().
[10]:
testDL.plot3D()
You appear to be running in JupyterLab (or JavaScript failed to load for some other reason). You need to install the 3dmol extension:
jupyter labextension install jupyterlab_3dmol
The molecule is stored as an RDkit object, self.mol, and RDkit methods are also available.
[11]:
type(testDL.mol)
[11]:
rdkit.Chem.rdchem.Mol
[11]:
# Calling the object will render it
testDL.mol
[11]:
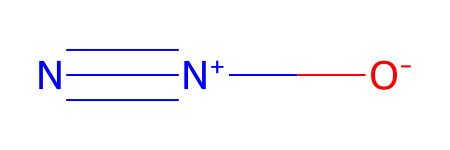
[12]:
# RDkit method example
testDL.mol.GetNumAtoms()
[12]:
3
pyGamess wrapper
Setup Gamess job
[14]:
# Init the pyGamess job.
# This minimally needs a gamess_path set, which defaults to '/opt/gamess'
testDL.initGamess() # Using defaults
*** Init pyGamess job.
Default Gamess input card set (use self.params to modify options dictionary, self.setGamess() to test):
$contrl scftyp=rhf runtyp=energy $end
$basis gbasis=sto ngauss=3 $end
$system mwords=30 $end
$DATA
None
C1
O 8.0 1.3063000000 0.0000000000 0.0000000000
N 7.0 -0.1096000000 0.0000000000 0.0000000000
N 7.0 -1.1967000000 0.0000000000 0.0000000000
$END
*** Found Gamess executable: /opt/gamess/gamess.00.x
This creates a pyGamess object, accessible at self.g, and properties can be inspected.
[16]:
print(type(testDL.g))
print(testDL.g.gamess_path)
<class 'epsman.elecStructure.gamess.gamessInput'>
/opt/gamess
All Gamess job parameters are stored in a dictionary, at self.params (also self.g.options), and can be set there directly using self.setParam() or dictionary methods. (For more details, see the pyGamess docs for more info, and the Gamess manual Input section for details of the available options.)
pyGamess doesn’t support symmetry or job annotation (uses ‘C1’ only), this can be added via a class wrapper here, and can be set via self.setGamess (this is also run automatically at class init). This adds an extras item to the params dictionary, with additional job details including symmetry.
Note that is symmetry is set to anything other than ‘C1’, coordinate transforms may be required. See notes below.
[18]:
# Show all params
testDL.params
[18]:
{'contrl': {'scftyp': 'rhf', 'runtyp': 'energy'},
'basis': {'gbasis': 'sto', 'ngauss': '3'},
'statpt': {'opttol': '0.0001', 'nstep': '20'},
'system': {'mwords': '30'},
'cis': {'nstate': '1'},
'extra': {'job': None, 'sym': 'C1', 'atomList': None}}
[19]:
# Params can be modified or added using the setParams method...
testDL.setParam(inputGroup='contrl',inputDict={'maxit':30})
Updating existing group 'contrl'. (To replace group, pass 'resetGroup=True')
Updated group 'contrl': {'scftyp': 'rhf', 'runtyp': 'energy', 'maxit': 30}
[20]:
# Pass resetGroup = True to replace all existing group settings with the passed dict (otherwise settings will be added)...
testDL.setParam(inputGroup='contrl',inputDict={'scftyp': 'rhf'}, resetGroup = True)
Replacing existing group 'contrl'.
Updated group 'contrl': {'scftyp': 'rhf'}
[21]:
# Params can also be modified or added using dictionary syntax
testDL.params['contrl']['runtyp']='energy'
testDL.params['contrl']['maxit'] = 60 # Add a control param. See https://www.msg.chem.iastate.edu/gamess/GAMESS_Manual/input.pdf
testDL.params
[21]:
{'contrl': {'scftyp': 'rhf', 'runtyp': 'energy', 'maxit': 60},
'basis': {'gbasis': 'sto', 'ngauss': '3'},
'statpt': {'opttol': '0.0001', 'nstep': '20'},
'system': {'mwords': '30'},
'cis': {'nstate': '1'},
'extra': {'job': None, 'sym': 'C1', 'atomList': None}}
The current Gamess input card can always be checked via self.printGamessInput()
[23]:
testDL.printGamessInput()
*** Gamess input card:
$contrl scftyp=rhf runtyp=energy maxit=60 $end
$basis gbasis=sto ngauss=3 $end
$system mwords=30 $end
$DATA
None
C1
O 8.0 1.3063000000 0.0000000000 0.0000000000
N 7.0 -0.1096000000 0.0000000000 0.0000000000
N 7.0 -1.1967000000 0.0000000000 0.0000000000
$END
Run Gamess
Basic energy run
If a valid gamess_path is set, then this is simple, and the basic results are returned to self.mol, along with some diagnostic information.
[25]:
# Run as per input card
testDL.runGamess()
INFO:pygamess.gamess:Executeing py_rungms with command /opt/gamess/ddikick.x /opt/gamess/gamess.00.x znjoyj -ddi 1 1 jake -scr /tmp/tmpah4dzgts > /tmp/tmpah4dzgts/znjoyj.out
*** ddikick exit status OK: ddikick.x: exited gracefully.
*** Gamess run completed OK.
E = -181.1830797993
[26]:
# Energy
testDL.E
[26]:
'-181.1830797993'
Run geometry optimization
Note this may fail in some cases for geometries not aligned in the preferred Gamess manner (major symmetry axis == z-axis), but in this case diagnostic info should be printed to screen.
[28]:
# Run optimization, in this case the updated coord table is also shown if self.verbose > 1
# testDL.verbose = 2
testDL.runGamess(runType = 'optimize')
INFO:pygamess.gamess:Executeing py_rungms with command /opt/gamess/ddikick.x /opt/gamess/gamess.00.x fdtcyj -ddi 1 1 jake -scr /tmp/tmpah4dzgts > /tmp/tmpah4dzgts/fdtcyj.out
*** Warning: found errors in Gamess output, type: Warnings
*** Check self.mol.GetProp('Warnings') for details.
*** Warning: found errors in Gamess output, type: ddikick
*** Check self.mol.GetProp('ddikick') for details.
*** Gamess run completed with warnings.
E = -181.1830797993
*** Gamess optimization run - reseting self.mol with updated coords.
Note that atom ordering may change depending on Gamess output.
Found 1 geometry iterations in Gamess output.
Updating with new coords, output set to self.mol
| Ind | Species | Atomic Num. | x | y | z | |
|---|---|---|---|---|---|---|
| 0 | 0 | O | 8 | 1.3063 | 0.0 | 0.0 |
| 1 | 1 | N | 7 | -0.1096 | 0.0 | 0.0 |
| 2 | 2 | N | 7 | -1.1967 | 0.0 | 0.0 |
*** WARNINGS FOUND IN GAMESS OUTPUT, values for E and molecular coords may reflect input molecule if run did not complete.
Set geom opt coord outputs to self.geomOpt.
Troubleshooting Gamess errors
In case of issues, the current outputs can be inspected (or maybe printed directly if self.verbose > 1).
[30]:
# Check errors if present
print(testDL.mol.GetProp('Warnings'))
print(testDL.mol.GetProp('ddikick'))
WARNING: EIGENVECTOR ROUTINE -EINVIT- DID NOT CONVERGE FOR VECTOR 3
WARNING: EIGENVECTOR ROUTINE -EINVIT- DID NOT CONVERGE FOR VECTOR 7
ddikick.x: application process 0 quit unexpectedly.
ddikick.x: Sending kill signal to DDI processes.
ddikick.x: Execution terminated due to error(s).
For geometry optimizations, the issue is typically the orientation of the coordinate system. A quick fix is to run the rotateFrame() method, which will try and orient the canonical RDkit alignment (symmetry axis == x-axis) to the canonical Gamess alignment (symmetry axis == z-axis), see below for more details. Note that basic energy runs usually work without this fix, but it is required for geometry optimization or use of symmetry.
[32]:
# A quick fix is to run the rotateFrame() method
testDL.rotateFrame()
testDL.verbose = 2 # If self.verbose > 1, runGamess will also print new (optimized) geom
testDL.runGamess(runType = 'optimize')
*** Updating coords (Pandas version).
*** Set frame rotations, new coord table:
| Ind | Species | Atomic Num. | x | y | z | |
|---|---|---|---|---|---|---|
| 0 | 0 | O | 8 | 0.0 | 0.0 | 1.3063 |
| 1 | 1 | N | 7 | 0.0 | 0.0 | -0.1096 |
| 2 | 2 | N | 7 | 0.0 | 0.0 | -1.1967 |
INFO:pygamess.gamess:Executeing py_rungms with command /opt/gamess/ddikick.x /opt/gamess/gamess.00.x jptncv -ddi 1 1 jake -scr /tmp/tmpah4dzgts > /tmp/tmpah4dzgts/jptncv.out
*** ddikick exit status OK: ddikick.x: exited gracefully.
*** Gamess run completed OK.
E = -181.2047216883
| Ind | Species | Atomic Num. | x | y | z | |
|---|---|---|---|---|---|---|
| 0 | 0 | O | 8 | 0.0 | 0.0 | 1.235127 |
| 1 | 1 | N | 7 | 0.0 | 0.0 | -0.040309 |
| 2 | 2 | N | 7 | 0.0 | 0.0 | -1.194818 |
*** Gamess optimization run - reseting self.mol with updated coords.
Note that atom ordering may change depending on Gamess output.
Found 9 geometry iterations in Gamess output.
Updated coords from Gamess run:
| Ind | Species | Atomic Num. | x | y | z | |
|---|---|---|---|---|---|---|
| 0 | 0 | O | 8 | 0.0 | 0.0 | 1.235127 |
| 1 | 1 | N | 7 | 0.0 | 0.0 | -0.040309 |
| 2 | 2 | N | 7 | 0.0 | 0.0 | -1.194818 |
Updating with new coords, output set to self.mol
| Ind | Species | Atomic Num. | x | y | z | |
|---|---|---|---|---|---|---|
| 0 | 0 | O | 8 | 0.0 | 0.0 | 1.235127 |
| 1 | 1 | N | 7 | 0.0 | 0.0 | -0.040309 |
| 2 | 2 | N | 7 | 0.0 | 0.0 | -1.194818 |
Updated coords from Gamess run:
| Ind | Species | Atomic Num. | x | y | z | ||
|---|---|---|---|---|---|---|---|
| Geom iter | Atom index | ||||||
| 0 | 0 | 0 | O | 8 | 0.0 | 0.0 | 1.306300 |
| 1 | 1 | N | 7 | 0.0 | 0.0 | -0.109600 | |
| 2 | 2 | N | 7 | 0.0 | 0.0 | -1.196700 | |
| 1 | 0 | 3 | O | 8 | 0.0 | 0.0 | 1.314860 |
| 1 | 4 | N | 7 | 0.0 | 0.0 | -0.023539 | |
| 2 | 5 | N | 7 | 0.0 | 0.0 | -1.291321 | |
| 2 | 0 | 6 | O | 8 | 0.0 | 0.0 | 1.282565 |
| 1 | 7 | N | 7 | 0.0 | 0.0 | -0.043055 | |
| 2 | 8 | N | 7 | 0.0 | 0.0 | -1.239510 | |
| 3 | 0 | 9 | O | 8 | 0.0 | 0.0 | 1.228473 |
| 1 | 10 | N | 7 | 0.0 | 0.0 | -0.047486 | |
| 2 | 11 | N | 7 | 0.0 | 0.0 | -1.180987 | |
| 4 | 0 | 12 | O | 8 | 0.0 | 0.0 | 1.232768 |
| 1 | 13 | N | 7 | 0.0 | 0.0 | -0.037089 | |
| 2 | 14 | N | 7 | 0.0 | 0.0 | -1.195679 | |
| 5 | 0 | 15 | O | 8 | 0.0 | 0.0 | 1.237216 |
| 1 | 16 | N | 7 | 0.0 | 0.0 | -0.041190 | |
| 2 | 17 | N | 7 | 0.0 | 0.0 | -1.196026 | |
| 6 | 0 | 18 | O | 8 | 0.0 | 0.0 | 1.234882 |
| 1 | 19 | N | 7 | 0.0 | 0.0 | -0.040325 | |
| 2 | 20 | N | 7 | 0.0 | 0.0 | -1.194557 | |
| 7 | 0 | 21 | O | 8 | 0.0 | 0.0 | 1.235127 |
| 1 | 22 | N | 7 | 0.0 | 0.0 | -0.040309 | |
| 2 | 23 | N | 7 | 0.0 | 0.0 | -1.194818 | |
| 8 | 0 | 24 | O | 8 | 0.0 | 0.0 | 1.235127 |
| 1 | 25 | N | 7 | 0.0 | 0.0 | -0.040309 | |
| 2 | 26 | N | 7 | 0.0 | 0.0 | -1.194818 |
Set geom opt coord outputs to self.geomOpt.
Gamess full output and log files
In the default case the tmp files are not kept. To keep the full Gamess output, supply a filename (full path).
The current output file path is set in self.gout.
[34]:
# Check for tmp file. This will always be set, but may have been deleted.
print(testDL.gout)
!cat {testDL.gout}
/tmp/tmpah4dzgts/tjweam.out
cat: /tmp/tmpah4dzgts/tjweam.out: No such file or directory
[35]:
# runGamess wrapper will take a path and move the output file.
testDL.runGamess(fileOut = '/tmp/test.out')
INFO:pygamess.gamess:Executeing py_rungms with command /opt/gamess/ddikick.x /opt/gamess/gamess.00.x qcvwnh -ddi 1 1 jake -scr /tmp/tmpah4dzgts > /tmp/tmpah4dzgts/qcvwnh.out
*** ddikick exit status OK: ddikick.x: exited gracefully.
*** Gamess run completed OK.
E = -181.2047216883
*** Gamess output file moved to /tmp/test.out
[36]:
# In this case the complete output file is retained, and can also be printed
print(testDL.gout)
testDL.printGamess()
/tmp/test.out
*** Contents from file /tmp/test.out.
Distributed Data Interface kickoff program.
Initiating 1 compute processes on 1 nodes to run the following command:
/opt/gamess/gamess.00.x qcvwnh
******************************************************
* GAMESS VERSION = 30 SEP 2018 (R3) *
* FROM IOWA STATE UNIVERSITY *
* M.W.SCHMIDT, K.K.BALDRIDGE, J.A.BOATZ, S.T.ELBERT, *
* M.S.GORDON, J.H.JENSEN, S.KOSEKI, N.MATSUNAGA, *
* K.A.NGUYEN, S.J.SU, T.L.WINDUS, *
* TOGETHER WITH M.DUPUIS, J.A.MONTGOMERY *
* J.COMPUT.CHEM. 14, 1347-1363(1993) *
**************** 64 BIT LINUX VERSION ****************
SINCE 1993, STUDENTS AND POSTDOCS WORKING AT IOWA STATE UNIVERSITY
AND ALSO IN THEIR VARIOUS JOBS AFTER LEAVING ISU HAVE MADE IMPORTANT
CONTRIBUTIONS TO THE CODE:
IVANA ADAMOVIC, CHRISTINE AIKENS, YURI ALEXEEV, POOJA ARORA,
ANDREY ASADCHEV, ROB BELL, PRADIPTA BANDYOPADHYAY, JONATHAN BENTZ,
BRETT BODE, KURT BRORSEN, CALEB CARLIN, GALINA CHABAN, WEI CHEN,
CHEOL HO CHOI, PAUL DAY, ALBERT DEFUSCO, NUWAN DESILVA, TIM DUDLEY,
DMITRI FEDOROV, ALEX FINDLATER, GRAHAM FLETCHER, MARK FREITAG,
KURT GLAESEMANN, ANASTASIA GUNINA,
DAN KEMP, GRANT MERRILL, NORIYUKI MINEZAWA, JONATHAN MULLIN,
TAKESHI NAGATA, SEAN NEDD, HEATHER NETZLOFF, BOSILJKA NJEGIC, RYAN OLSON,
MIKE PAK, BUU PHAM,
SPENCER PRUITT, LUKE ROSKOP, JIM SHOEMAKER, LYUDMILA SLIPCHENKO,
TONY SMITH, SAROM SOK LEANG, JIE SONG, TETSUYA TAKETSUGU, SIMON WEBB,
PENG XU, SOOHAENG YOO, FEDERICO ZAHARIEV
ADDITIONAL CODE HAS BEEN PROVIDED BY COLLABORATORS IN OTHER GROUPS:
IOWA STATE UNIVERSITY:
JOE IVANIC, AARON WEST, LAIMUTIS BYTAUTAS, KLAUS RUEDENBERG
UNIVERSITY OF TOKYO: KIMIHIKO HIRAO, TAKAHITO NAKAJIMA,
TAKAO TSUNEDA, MUNEAKI KAMIYA, SUSUMU YANAGISAWA,
KIYOSHI YAGI, MAHITO CHIBA, SEIKEN TOKURA, NAOAKI KAWAKAMI
UNIVERSITY OF AARHUS: FRANK JENSEN
UNIVERSITY OF IOWA: VISVALDAS KAIRYS, HUI LI
NATIONAL INST. OF STANDARDS AND TECHNOLOGY: WALT STEVENS, DAVID GARMER
UNIVERSITY OF PISA: BENEDETTA MENNUCCI, JACOPO TOMASI
UNIVERSITY OF MEMPHIS: HENRY KURTZ, PRAKASHAN KORAMBATH
UNIVERSITY OF ALBERTA: TOBY ZENG, MARIUSZ KLOBUKOWSKI
UNIVERSITY OF NEW ENGLAND: MARK SPACKMAN
MIE UNIVERSITY: HIROAKI UMEDA
NAT. INST. OF ADVANCED INDUSTRIAL SCIENCE AND TECHNOLOGY: KAZUO KITAURA
MICHIGAN STATE UNIVERSITY:
KAROL KOWALSKI, MARTA WLOCH, JEFFREY GOUR, JESSE LUTZ,
WEI LI, JUN SHEN, J. EMILIANO DEUSTUA, PIOTR PIECUCH
UNIVERSITY OF MINNESOTA:
YINAN SHU
UNIVERSITY OF SILESIA: MONIKA MUSIAL, STANISLAW KUCHARSKI
FACULTES UNIVERSITAIRES NOTRE-DAME DE LA PAIX:
OLIVIER QUINET, BENOIT CHAMPAGNE
UNIVERSITY OF CALIFORNIA - SANTA BARBARA: BERNARD KIRTMAN
INSTITUTE FOR MOLECULAR SCIENCE:
KAZUYA ISHIMURA, MICHIO KATOUDA, AND SHIGERU NAGASE
UNIVERSITY OF NOTRE DAME: ANNA POMOGAEVA, DAN CHIPMAN
KYUSHU UNIVERSITY:
HARUYUKI NAKANO,
FENG LONG GU, JACEK KORCHOWIEC, MARCIN MAKOWSKI, AND YURIKO AOKI,
HIROTOSHI MORI AND EISAKU MIYOSHI
PENNSYLVANIA STATE UNIVERSITY:
TZVETELIN IORDANOV, CHET SWALINA, JONATHAN SKONE,
SHARON HAMMES-SCHIFFER
WASEDA UNIVERSITY:
MASATO KOBAYASHI, TOMOKO AKAMA, TSUGUKI TOUMA,
TAKESHI YOSHIKAWA, YASUHIRO IKABATA, JUNJI SEINO,
YUYA NAKAJIMA, HIROMI NAKAI
NANJING UNIVERSITY: SHUHUA LI
UNIVERSITY OF NEBRASKA:
PEIFENG SU, DEJUN SI, NANDUN THELLAMUREGE, YALI WANG, HUI LI
UNIVERSITY OF ZURICH: ROBERTO PEVERATI, KIM BALDRIDGE
N. COPERNICUS UNIVERSITY AND JACKSON STATE UNIVERSITY:
MARIA BARYSZ
UNIVERSITY OF COPENHAGEN: Jimmy Kromann, CASPER STEINMANN
TOKYO INSTITUTE OF TECHNOLOGY: HIROYA NAKATA
NAGOYA UNIVERSITY: YOSHIO NISHIMOTO, STEPHAN IRLE
MOSCOW STATE UNIVERSITY: VLADIMIR MIRONOV
EXECUTION OF GAMESS BEGUN Thu Jan 25 14:15:21 2024
ECHO OF THE FIRST FEW INPUT CARDS -
INPUT CARD> $contrl scftyp=rhf runtyp=energy maxit=60 $end
INPUT CARD> $basis gbasis=sto ngauss=3 $end
INPUT CARD> $system mwords=30 $end
INPUT CARD> $DATA
INPUT CARD>None
INPUT CARD>C1
INPUT CARD>O 8.0 0.0000000000 0.0000000000 1.2351270000
INPUT CARD>N 7.0 0.0000000000 0.0000000000 -0.0403090000
INPUT CARD>N 7.0 0.0000000000 0.0000000000 -1.1948180000
INPUT CARD> $END
30000000 WORDS OF MEMORY AVAILABLE
BASIS OPTIONS
-------------
GBASIS=STO IGAUSS= 3 POLAR=NONE
NDFUNC= 0 NFFUNC= 0 DIFFSP= F
NPFUNC= 0 DIFFS= F BASNAM=
RUN TITLE
---------
None
THE POINT GROUP OF THE MOLECULE IS C1
THE ORDER OF THE PRINCIPAL AXIS IS 0
ATOM ATOMIC COORDINATES (BOHR)
CHARGE X Y Z
O 8.0 0.0000000000 0.0000000000 2.3340515900
N 7.0 0.0000000000 0.0000000000 -0.0761729648
N 7.0 0.0000000000 0.0000000000 -2.2578786252
INTERNUCLEAR DISTANCES (ANGS.)
------------------------------
1 O 2 N 3 N
1 O 0.0000000 1.2754360 * 2.4299450 *
2 N 1.2754360 * 0.0000000 1.1545090 *
3 N 2.4299450 * 1.1545090 * 0.0000000
* ... LESS THAN 3.000
ATOMIC BASIS SET
----------------
THE CONTRACTED PRIMITIVE FUNCTIONS HAVE BEEN UNNORMALIZED
THE CONTRACTED BASIS FUNCTIONS ARE NOW NORMALIZED TO UNITY
SHELL TYPE PRIMITIVE EXPONENT CONTRACTION COEFFICIENT(S)
O
1 S 1 130.7093214 0.154328967295
1 S 2 23.8088661 0.535328142282
1 S 3 6.4436083 0.444634542185
2 L 4 5.0331513 -0.099967229187 0.155916274999
2 L 5 1.1695961 0.399512826089 0.607683718598
2 L 6 0.3803890 0.700115468880 0.391957393099
N
3 S 7 99.1061690 0.154328967295
3 S 8 18.0523124 0.535328142282
3 S 9 4.8856602 0.444634542185
4 L 10 3.7804559 -0.099967229187 0.155916274999
4 L 11 0.8784966 0.399512826089 0.607683718598
4 L 12 0.2857144 0.700115468880 0.391957393099
N
5 S 13 99.1061690 0.154328967295
5 S 14 18.0523124 0.535328142282
5 S 15 4.8856602 0.444634542185
6 L 16 3.7804559 -0.099967229187 0.155916274999
6 L 17 0.8784966 0.399512826089 0.607683718598
6 L 18 0.2857144 0.700115468880 0.391957393099
TOTAL NUMBER OF BASIS SET SHELLS = 6
NUMBER OF CARTESIAN GAUSSIAN BASIS FUNCTIONS = 15
NUMBER OF ELECTRONS = 22
CHARGE OF MOLECULE = 0
SPIN MULTIPLICITY = 1
NUMBER OF OCCUPIED ORBITALS (ALPHA) = 11
NUMBER OF OCCUPIED ORBITALS (BETA ) = 11
TOTAL NUMBER OF ATOMS = 3
THE NUCLEAR REPULSION ENERGY IS 57.8891485376
THIS MOLECULE IS RECOGNIZED AS BEING LINEAR,
ORBITAL LZ DEGENERACY TOLERANCE ETOLLZ= 1.00E-06
$CONTRL OPTIONS
---------------
SCFTYP=RHF RUNTYP=ENERGY EXETYP=RUN
MPLEVL= 0 CITYP =NONE CCTYP =NONE VBTYP =NONE
DFTTYP=NONE TDDFT =NONE
MULT = 1 ICHARG= 0 NZVAR = 0 COORD =UNIQUE
PP =NONE RELWFN=NONE LOCAL =NONE NUMGRD= F
ISPHER= -1 NOSYM = 0 MAXIT = 60 UNITS =ANGS
PLTORB= F MOLPLT= F AIMPAC= F FRIEND=
NPRINT= 7 IREST = 0 GEOM =INPUT
NORMF = 0 NORMP = 0 ITOL = 20 ICUT = 9
INTTYP=BEST GRDTYP=BEST QMTTOL= 1.0E-06
$SYSTEM OPTIONS
---------------
REPLICATED MEMORY= 30000000 WORDS (ON EVERY NODE).
DISTRIBUTED MEMDDI= 0 MILLION WORDS IN AGGREGATE,
MEMDDI DISTRIBUTED OVER 1 PROCESSORS IS 0 WORDS/PROCESSOR.
TOTAL MEMORY REQUESTED ON EACH PROCESSOR= 30000000 WORDS.
TIMLIM= 525600.00 MINUTES, OR 365.0 DAYS.
PARALL= F BALTYP= DLB KDIAG= 0 COREFL= F
MXSEQ2= 300 MXSEQ3= 150 mem10= 0
----------------
PROPERTIES INPUT
----------------
MOMENTS FIELD POTENTIAL DENSITY
IEMOM = 1 IEFLD = 0 IEPOT = 0 IEDEN = 0
WHERE =COMASS WHERE =NUCLEI WHERE =NUCLEI WHERE =NUCLEI
OUTPUT=BOTH OUTPUT=BOTH OUTPUT=BOTH OUTPUT=BOTH
IEMINT= 0 IEFINT= 0 IEDINT= 0
MORB = 0
EXTRAPOLATION IN EFFECT
SOSCF IN EFFECT
ORBITAL PRINTING OPTION: NPREO= 1 15 2 1
-------------------------------
INTEGRAL TRANSFORMATION OPTIONS
-------------------------------
NWORD = 0
CUTOFF = 1.0E-09 MPTRAN = 0
DIRTRF = F AOINTS =DUP
----------------------
INTEGRAL INPUT OPTIONS
----------------------
NOPK = 1 NORDER= 0 SCHWRZ= F
------------------------------------------
THE POINT GROUP IS C1 , NAXIS= 0, ORDER= 1
------------------------------------------
DIMENSIONS OF THE SYMMETRY SUBSPACES ARE
A = 15
..... DONE SETTING UP THE RUN .....
STEP CPU TIME = 0.00 TOTAL CPU TIME = 0.0 ( 0.0 MIN)
TOTAL WALL CLOCK TIME= 0.0 SECONDS, CPU UTILIZATION IS 100.00%
********************
1 ELECTRON INTEGRALS
********************
...... END OF ONE-ELECTRON INTEGRALS ......
STEP CPU TIME = 0.01 TOTAL CPU TIME = 0.0 ( 0.0 MIN)
TOTAL WALL CLOCK TIME= 0.0 SECONDS, CPU UTILIZATION IS 100.00%
-------------
GUESS OPTIONS
-------------
GUESS =HUCKEL NORB = 0 NORDER= 0
MIX = F PRTMO = F PUNMO = F
TOLZ = 1.0E-08 TOLE = 1.0E-05
SYMDEN= F PURIFY= F
INITIAL GUESS ORBITALS GENERATED BY HUCKEL ROUTINE.
HUCKEL GUESS REQUIRES 4764 WORDS.
SYMMETRIES FOR INITIAL GUESS ORBITALS FOLLOW. BOTH SET(S).
11 ORBITALS ARE OCCUPIED ( 3 CORE ORBITALS).
4=A 5=A 6=A 7=A 8=A 9=A 10=A
11=A 12=A 13=A 14=A 15=A
...... END OF INITIAL ORBITAL SELECTION ......
STEP CPU TIME = 0.00 TOTAL CPU TIME = 0.0 ( 0.0 MIN)
TOTAL WALL CLOCK TIME= 0.0 SECONDS, CPU UTILIZATION IS 100.00%
----------------------
AO INTEGRAL TECHNOLOGY
----------------------
S,P,L SHELL ROTATED AXIS INTEGRALS, REPROGRAMMED BY
KAZUYA ISHIMURA (IMS) AND JOSE SIERRA (SYNSTAR).
S,P,D,L SHELL ROTATED AXIS INTEGRALS PROGRAMMED BY
KAZUYA ISHIMURA (INSTITUTE FOR MOLECULAR SCIENCE).
S,P,D,F,G SHELL TO TOTAL QUARTET ANGULAR MOMENTUM SUM 5,
ERIC PROGRAM BY GRAHAM FLETCHER (ELORET AND NASA ADVANCED
SUPERCOMPUTING DIVISION, AMES RESEARCH CENTER).
S,P,D,F,G,L SHELL GENERAL RYS QUADRATURE PROGRAMMED BY
MICHEL DUPUIS (PACIFIC NORTHWEST NATIONAL LABORATORY).
--------------------
2 ELECTRON INTEGRALS
--------------------
THE -PK- OPTION IS OFF, THE INTEGRALS ARE NOT IN SUPERMATRIX FORM.
STORING 15000 INTEGRALS/RECORD ON DISK, USING 12 BYTES/INTEGRAL.
TWO ELECTRON INTEGRAL EVALUATION REQUIRES 89377 WORDS OF MEMORY.
II,JST,KST,LST = 1 1 1 1 NREC = 1 INTLOC = 1
II,JST,KST,LST = 2 1 1 1 NREC = 1 INTLOC = 2
II,JST,KST,LST = 3 1 1 1 NREC = 1 INTLOC = 34
II,JST,KST,LST = 4 1 1 1 NREC = 1 INTLOC = 80
II,JST,KST,LST = 5 1 1 1 NREC = 1 INTLOC = 506
II,JST,KST,LST = 6 1 1 1 NREC = 1 INTLOC = 729
TOTAL NUMBER OF NONZERO TWO-ELECTRON INTEGRALS = 2309
1 INTEGRAL RECORDS WERE STORED ON DISK FILE 8.
...... END OF TWO-ELECTRON INTEGRALS .....
STEP CPU TIME = 0.01 TOTAL CPU TIME = 0.0 ( 0.0 MIN)
TOTAL WALL CLOCK TIME= 0.0 SECONDS, CPU UTILIZATION IS 100.00%
--------------------------
RHF SCF CALCULATION
--------------------------
NUCLEAR ENERGY = 57.8891485376
MAXIT = 60 NPUNCH= 2
EXTRAP=T DAMP=F SHIFT=F RSTRCT=F DIIS=F DEM=F SOSCF=T
DENSITY MATRIX CONV= 1.00E-05
SOSCF WILL OPTIMIZE 44 ORBITAL ROTATIONS, SOGTOL= 0.250
MEMORY REQUIRED FOR RHF ITERS= 31961 WORDS.
ITER EX DEM TOTAL ENERGY E CHANGE DENSITY CHANGE ORB. GRAD
1 0 0 -180.7238079779 -180.7238079779 0.660736387 0.000000000
---------------START SECOND ORDER SCF---------------
2 1 0 -181.0836015402 -0.3597935624 0.427865508 0.159013058
3 2 0 -181.1160394403 -0.0324379001 0.243273093 0.126000619
4 3 0 -181.1957291392 -0.0796896989 0.127989704 0.047620555
5 4 0 -181.2016339913 -0.0059048521 0.040297486 0.025416306
6 5 0 -181.2045894393 -0.0029554481 0.009025426 0.002820831
7 6 0 -181.2047002250 -0.0001107857 0.006398880 0.001328239
8 7 0 -181.2047214849 -0.0000212599 0.000413802 0.000143818
9 8 0 -181.2047216547 -0.0000001698 0.000298380 0.000051251
10 9 0 -181.2047216879 -0.0000000332 0.000027016 0.000014773
11 10 0 -181.2047216882 -0.0000000003 0.000006117 0.000002884
12 11 0 -181.2047216883 -0.0000000000 0.000000752 0.000000233
-----------------
DENSITY CONVERGED
-----------------
TIME TO FORM FOCK OPERATORS= 0.0 SECONDS ( 0.0 SEC/ITER)
TIME TO SOLVE SCF EQUATIONS= 0.0 SECONDS ( 0.0 SEC/ITER)
FINAL RHF ENERGY IS -181.2047216883 AFTER 12 ITERATIONS
LZ VALUE ANALYSIS FOR THE MOS
----------------------------------------
MO 1 ( 1) HAS LZ(WEIGHT)= 0.00(100.0%)
MO 2 ( 2) HAS LZ(WEIGHT)= 0.00(100.0%)
MO 3 ( 3) HAS LZ(WEIGHT)= 0.00(100.0%)
MO 4 ( 4) HAS LZ(WEIGHT)= 0.00(100.0%)
MO 5 ( 5) HAS LZ(WEIGHT)= 0.00(100.0%)
MO 6 ( 6) HAS LZ(WEIGHT)= 0.00(100.0%)
MO 7 ( 7) HAS LZ(WEIGHT)=-1.00( 50.0%) 1.00( 50.0%)
MO 8 ( 7) HAS LZ(WEIGHT)=-1.00( 50.0%) 1.00( 50.0%)
MO 9 ( 8) HAS LZ(WEIGHT)= 0.00(100.0%)
MO 10 ( 9) HAS LZ(WEIGHT)=-1.00( 50.0%) 1.00( 50.0%)
MO 11 ( 9) HAS LZ(WEIGHT)=-1.00( 50.0%) 1.00( 50.0%)
MO 12 ( 10) HAS LZ(WEIGHT)=-1.00( 50.0%) 1.00( 50.0%)
MO 13 ( 10) HAS LZ(WEIGHT)=-1.00( 50.0%) 1.00( 50.0%)
MO 14 ( 11) HAS LZ(WEIGHT)= 0.00(100.0%)
MO 15 ( 12) HAS LZ(WEIGHT)= 0.00(100.0%)
------------
EIGENVECTORS
------------
1 2 3 4 5
-20.3101 -15.6684 -15.5054 -1.4784 -1.3050
A A A A A
1 O 1 S 0.994521 0.000368 -0.000002 -0.112063 -0.175539
2 O 1 S 0.023722 -0.003860 -0.000081 0.382992 0.617717
3 O 1 X -0.000000 -0.000000 0.000000 -0.000000 0.000000
4 O 1 Y -0.000000 -0.000000 0.000000 -0.000000 0.000000
5 O 1 Z -0.004395 0.002307 0.000065 -0.104386 -0.087235
6 N 2 S 0.000420 0.994008 -0.002574 -0.190648 0.020098
7 N 2 S -0.004554 0.027883 -0.008861 0.573738 -0.041247
8 N 2 X -0.000000 -0.000000 0.000000 -0.000000 0.000000
9 N 2 Y -0.000000 -0.000000 0.000000 -0.000000 0.000000
10 N 2 Z -0.005390 -0.002942 0.005500 -0.060099 0.397917
11 N 3 S 0.000006 0.003133 0.994207 -0.120348 0.128087
12 N 3 S -0.000877 -0.007170 0.027020 0.340323 -0.367292
13 N 3 X -0.000000 -0.000000 0.000000 -0.000000 0.000000
14 N 3 Y -0.000000 -0.000000 0.000000 -0.000000 0.000000
15 N 3 Z -0.000640 -0.004210 0.007705 0.163870 -0.133984
6 7 8 9 10
-0.7289 -0.6620 -0.6620 -0.5855 -0.3560
A A A A A
1 O 1 S -0.139872 -0.000000 0.000000 0.076373 -0.000000
2 O 1 S 0.651573 -0.000000 0.000000 -0.389374 -0.000000
3 O 1 X 0.000000 0.305187 -0.193924 -0.000000 0.196032
4 O 1 Y 0.000000 0.193924 0.305187 -0.000000 0.821561
5 O 1 Z 0.322387 -0.000000 0.000000 -0.328698 -0.000000
6 N 2 S 0.136535 -0.000000 0.000000 0.003394 -0.000000
7 N 2 S -0.597056 -0.000000 0.000000 0.014496 -0.000000
8 N 2 X 0.000000 0.576401 -0.366261 -0.000000 -0.022612
9 N 2 Y 0.000000 0.366261 0.576401 -0.000000 -0.094764
10 N 2 Z -0.198325 -0.000000 0.000000 0.480332 -0.000000
11 N 3 S -0.139942 -0.000000 0.000000 -0.118990 -0.000000
12 N 3 S 0.603592 -0.000000 0.000000 0.621231 -0.000000
13 N 3 X 0.000000 0.356087 -0.226267 -0.000000 -0.123512
14 N 3 Y 0.000000 0.226267 0.356087 -0.000000 -0.517632
15 N 3 Z -0.206288 -0.000000 0.000000 -0.515008 -0.000000
11 12 13 14 15
-0.3560 0.2266 0.2266 0.4956 1.1292
A A A A A
1 O 1 S -0.000000 0.000000 0.000000 0.068527 0.061891
2 O 1 S -0.000000 0.000000 0.000000 -0.407452 -0.434423
3 O 1 X 0.821561 -0.107211 -0.414072 -0.000000 -0.000000
4 O 1 Y -0.196032 -0.414072 0.107211 -0.000000 -0.000000
5 O 1 Z -0.000000 0.000000 0.000000 0.881539 0.563952
6 N 2 S -0.000000 0.000000 0.000000 -0.163174 0.050716
7 N 2 S -0.000000 0.000000 0.000000 1.117036 -0.559477
8 N 2 X -0.094764 0.197370 0.762288 -0.000000 -0.000000
9 N 2 Y 0.022612 0.762288 -0.197370 -0.000000 -0.000000
10 N 2 Z -0.000000 0.000000 0.000000 0.123196 1.363065
11 N 3 S -0.000000 0.000000 0.000000 0.066841 -0.104770
12 N 3 S -0.000000 0.000000 0.000000 -0.432367 0.945314
13 N 3 X -0.517632 -0.195308 -0.754327 -0.000000 -0.000000
14 N 3 Y 0.123512 -0.754327 0.195308 -0.000000 -0.000000
15 N 3 Z -0.000000 0.000000 0.000000 -0.639169 1.012984
...... END OF RHF CALCULATION ......
STEP CPU TIME = 0.00 TOTAL CPU TIME = 0.0 ( 0.0 MIN)
TOTAL WALL CLOCK TIME= 0.0 SECONDS, CPU UTILIZATION IS 100.00%
----------------------------------------------------------------
PROPERTY VALUES FOR THE RHF SELF-CONSISTENT FIELD WAVEFUNCTION
----------------------------------------------------------------
-----------------
ENERGY COMPONENTS
-----------------
WAVEFUNCTION NORMALIZATION = 1.0000000000
ONE ELECTRON ENERGY = -362.9526476470
TWO ELECTRON ENERGY = 123.8587774211
NUCLEAR REPULSION ENERGY = 57.8891485376
------------------
TOTAL ENERGY = -181.2047216883
ELECTRON-ELECTRON POTENTIAL ENERGY = 123.8587774211
NUCLEUS-ELECTRON POTENTIAL ENERGY = -542.0062674089
NUCLEUS-NUCLEUS POTENTIAL ENERGY = 57.8891485376
------------------
TOTAL POTENTIAL ENERGY = -360.2583414502
TOTAL KINETIC ENERGY = 179.0536197619
VIRIAL RATIO (V/T) = 2.0120137305
...... PI ENERGY ANALYSIS ......
ENERGY ANALYSIS:
FOCK ENERGY= -115.2350930820
BARE H ENERGY= -362.9526476470
ELECTRONIC ENERGY = -239.0938703645
KINETIC ENERGY= 179.0536197619
N-N REPULSION= 57.8891485376
TOTAL ENERGY= -181.2047218269
SIGMA PART(1+2)= -195.1428709933
(K,V1,2)= 162.9512486081 -442.0736108680 83.9794912666
PI PART(1+2)= -43.9509993712
(K,V1,2)= 16.1023711538 -99.9326565409 39.8792860159
SIGMA SKELETON, ERROR= -137.2537224557 0.0000000000
MIXED PART= 0.00000E+00 0.00000E+00 0.00000E+00 0.00000E+00
...... END OF PI ENERGY ANALYSIS ......
---------------------------------------
MULLIKEN AND LOWDIN POPULATION ANALYSES
---------------------------------------
ATOMIC MULLIKEN POPULATION IN EACH MOLECULAR ORBITAL
1 2 3 4 5
2.000000 2.000000 2.000000 2.000000 2.000000
1 2.000773 -0.000397 -0.000000 0.448171 0.895684
2 -0.000771 2.001775 -0.001738 1.017968 0.660247
3 -0.000002 -0.001378 2.001738 0.533861 0.444069
6 7 8 9 10
2.000000 2.000000 2.000000 2.000000 2.000000
1 0.855069 0.341908 0.341908 0.469754 1.394707
2 0.483611 1.155625 1.155625 0.349136 0.018853
3 0.661319 0.502467 0.502467 1.181110 0.586440
11
2.000000
1 1.394707
2 0.018853
3 0.586440
----- POPULATIONS IN EACH AO -----
MULLIKEN LOWDIN
1 O 1 S 1.99879 1.99799
2 O 1 S 1.91465 1.86685
3 O 1 X 1.73661 1.73801
4 O 1 Y 1.73661 1.73801
5 O 1 Z 0.75562 0.84110
6 N 2 S 1.99649 1.99278
7 N 2 S 1.40149 1.30498
8 N 2 X 1.17448 1.16636
9 N 2 Y 1.17448 1.16636
10 N 2 Z 1.11225 1.12483
11 N 3 S 1.99786 1.99704
12 N 3 S 1.77667 1.69853
13 N 3 X 1.08891 1.09562
14 N 3 Y 1.08891 1.09562
15 N 3 Z 1.04619 1.17590
----- MULLIKEN ATOMIC OVERLAP POPULATIONS -----
(OFF-DIAGONAL ELEMENTS NEED TO BE MULTIPLIED BY 2)
1 2 3
1 7.9278182
2 0.2255980 6.0773158
3 -0.0111331 0.5562713 6.4533936
TOTAL MULLIKEN AND LOWDIN ATOMIC POPULATIONS
ATOM MULL.POP. CHARGE LOW.POP. CHARGE
1 O 8.142283 -0.142283 8.181970 -0.181970
2 N 6.859185 0.140815 6.755318 0.244682
3 N 6.998532 0.001468 7.062712 -0.062712
-------------------------------
BOND ORDER AND VALENCE ANALYSIS BOND ORDER THRESHOLD=0.050
-------------------------------
BOND BOND BOND
ATOM PAIR DIST ORDER ATOM PAIR DIST ORDER ATOM PAIR DIST ORDER
1 2 1.275 1.307 1 3 2.430 0.516 2 3 1.155 2.453
TOTAL BONDED FREE
ATOM VALENCE VALENCE VALENCE
1 O 1.823 1.823 0.000
2 N 3.760 3.760 0.000
3 N 2.969 2.969 0.000
---------------------
ELECTROSTATIC MOMENTS
---------------------
POINT 1 X Y Z (BOHR) CHARGE
0.000000 0.000000 0.105658 -0.00 (A.U.)
DX DY DZ /D/ (DEBYE)
0.000000 0.000000 -0.489412 0.489412
...... END OF PROPERTY EVALUATION ......
STEP CPU TIME = 0.00 TOTAL CPU TIME = 0.0 ( 0.0 MIN)
TOTAL WALL CLOCK TIME= 0.0 SECONDS, CPU UTILIZATION IS 100.00%
580000 WORDS OF DYNAMIC MEMORY USED
EXECUTION OF GAMESS TERMINATED NORMALLY Thu Jan 25 14:15:21 2024
DDI: 263640 bytes (0.3 MB / 0 MWords) used by master data server.
----------------------------------------
CPU timing information for all processes
========================================
0: 0.27 + 0.00 = 0.27
----------------------------------------
ddikick.x: exited gracefully.
[37]:
# For quick checks, there are also head and tail functions
testDL.tail()
*** Contents from file /tmp/test.out.
Showing 20 tail lines.
ELECTROSTATIC MOMENTS
---------------------
POINT 1 X Y Z (BOHR) CHARGE
0.000000 0.000000 0.105658 -0.00 (A.U.)
DX DY DZ /D/ (DEBYE)
0.000000 0.000000 -0.489412 0.489412
...... END OF PROPERTY EVALUATION ......
STEP CPU TIME = 0.00 TOTAL CPU TIME = 0.0 ( 0.0 MIN)
TOTAL WALL CLOCK TIME= 0.0 SECONDS, CPU UTILIZATION IS 100.00%
580000 WORDS OF DYNAMIC MEMORY USED
EXECUTION OF GAMESS TERMINATED NORMALLY Thu Jan 25 14:15:21 2024
DDI: 263640 bytes (0.3 MB / 0 MWords) used by master data server.
----------------------------------------
CPU timing information for all processes
========================================
0: 0.27 + 0.00 = 0.27
----------------------------------------
ddikick.x: exited gracefully.
Symmetry & frame transformations
To use symmetry in the Gamess calculations, the system must be oriented such that the Z-axis is the highest symmetry axis. In tests both PubChem and RDkit seem to use the X-axis as the symmetry axis, so the frame needs to be rotated in general.
For more details, seethe symmetry-focussed docs.
From the Gamess manual:
The 'master frame' is just a standard orientation for
the molecule. By default, the 'master frame' assumes that
1. z is the principal rotation axis (if any),
2. x is a perpendicular two-fold axis (if any),
3. xz is the sigma-v plane (if any), and
4. xy is the sigma-h plane (if any).
Use the lowest number rule that applies to your molecule.
Some examples of these rules:
Ammonia (C3v): the unique H lies in the XZ plane (R1,R3).
Ethane (D3d): the unique H lies in the YZ plane (R1,R2).
Methane (Td): the H lies in the XYZ direction (R2). Since
there is more than one 3-fold, R1 does not apply.
HP=O (Cs): the mirror plane is the XY plane (R4).
In general, it is a poor idea to try to reorient the
molecule. Certain sections of the program, such as the
orbital symmetry assignment, do not know how to deal with
cases where the 'master frame' has been changed.
Linear molecules (C4v or D4h) must lie along the z axis,
so do not try to reorient linear molecules.
This is set with self.rotateFrame(), which can set arbitrary rotations, but defaults to X > Z axis transformation. This uses the RDkit canoicalise and transformation functions, with rotation matrices, as per Github user iwatobipen’s example notebook. (Thanks to iwatobipen and the RDkit list.)
[38]:
# Rotate the frame - default should align to Z-axis
testDL.rotateFrame()
*** Updating coords (Pandas version).
*** Set frame rotations, new coord table:
| Ind | Species | Atomic Num. | x | y | z | |
|---|---|---|---|---|---|---|
| 0 | 0 | O | 8 | 0.0 | 0.0 | 1.235127 |
| 1 | 1 | N | 7 | 0.0 | 0.0 | -0.040309 |
| 2 | 2 | N | 7 | 0.0 | 0.0 | -1.194818 |
Symmetry groups supported (from the Gamess manual):
GROUP is the Schoenflies symbol of the symmetry group,
you may choose from
C1, Cs, Ci, Cn, S2n, Cnh, Cnv, Dn, Dnh, Dnd,
T, Th, Td, O, Oh.
NAXIS is the order of the highest rotation axis, and
must be given when the name of the group contains an N.
For example, "Cnv 2" is C2v. "S2n 3" means S6. Use of
NAXIS up to 8 is supported in each axial groups.
For linear molecules, choose either Cnv or Dnh, and enter
NAXIS as 4. Enter atoms as Dnh with NAXIS=2. If the
electronic state of either is degenerate, check the note
about the effect of symmetry in the electronic state
in the SCF section of REFS.DOC.
[39]:
# Set Gamess input with symmetry
testDL.setGamess(note='N2O sym testing', sym='CNV 8')
Set sym = CNV 8
*** Gamess input card:
$contrl scftyp=rhf runtyp=energy maxit=60 $end
$basis gbasis=sto ngauss=3 $end
$system mwords=30 $end
$DATA
N2O sym testing
CNV 8
O 8.0 0.0000000000 0.0000000000 1.2351270000
N 7.0 0.0000000000 0.0000000000 -0.0403090000
N 7.0 0.0000000000 0.0000000000 -1.1948180000
$END
Finally, it is worth noting that symmetrized jobs require only the unique atoms given on the input card. This is currently accomplished here rather crudely, via a list of atom indices (rows) to the input builder.
For example…
[40]:
testDL.setGamess(note='N2O sym testing', sym='CNV 8', atomList = [0,2])
Set sym = CNV 8
Set atomList = [0, 2]
*** Gamess input card:
$contrl scftyp=rhf runtyp=energy maxit=60 $end
$basis gbasis=sto ngauss=3 $end
$system mwords=30 $end
$DATA
N2O sym testing
CNV 8
O 8.0 0.0000000000 0.0000000000 1.2351270000
N 7.0 0.0000000000 0.0000000000 -1.1948180000
$END
Where self.params['extra']['atomList'] gives the sub-selection on which atoms are listed on the input card for symmetrized jobs (TODO: make this better/automated!).
[41]:
testDL.params['extra']
[41]:
{'job': 'N2O sym testing', 'sym': 'CNV 8', 'atomList': [0, 2]}
For more details, seethe symmetry-focussed docs.
Additional Gamess parameters
For the full set of Gamess input options (inc. basis sets), see the manual.
As per the above input cards, pyGamess writes only the minimal set of $contrl, $basis and $system inputs, using the supplied parameters dictionary.
For setting the basis set, a helper method self.setBasis() can be used. For general parameter setting, self.setParam() can be used, along with the specific Gamess group name for the paramter.
[42]:
# The setBasis method wraps the PyGamess basis_set() method
testDL.setBasis("6-31G**")
Set basis to specification 6-31G**.
self.params['basis']: {'gbasis': 'N31', 'ngauss': '6', 'ndfunc': '1', 'npfunc': '1'}
[43]:
# This only has limited support...
testDL.setBasis("ACCD")
basis type not found
basis type not found
ERROR:pygamess.gamess:basis type not found
*** Basis configuration ACCD not supported by PyGamess.
To set manually, pass Gamess basis params as a dictionary to self.setParam().
E.g. for 'ACCD' configure with self.setParam(inputGroup='basis',inputDict={'gbasis':'ACCD'}),
Any other required params can also be set, e.g. self.setParam(inputGroup='contrl',inputDict={'ISPHER':'1'}).
See the Gamess manual for settings, https://www.msg.chem.iastate.edu/gamess/GAMESS_Manual/docs-input.txt.
[44]:
# ... applying settings manually will always work however
testDL.setParam(inputGroup='basis',inputDict={'gbasis':'ACCD'}, resetGroup=True) # Pass resetGroup=True to replace existing.
testDL.setParam(inputGroup='contrl',inputDict={'ISPHER':'1'})
# Note that dictionary syntax also works here, e.g. the above can also be set via:
# testDL.params['basis'] = {'gbasis':'ACCD'}
# testDL.params['contrl']['ISPHER']='1' # For ACCD need this too!
#
# But dictionary style will NOT work for 'extra' items.
Replacing existing group 'basis'.
Updated group 'basis': {'gbasis': 'ACCD'}
Updating existing group 'contrl'. (To replace group, pass 'resetGroup=True')
Updated group 'contrl': {'scftyp': 'rhf', 'runtyp': 'energy', 'maxit': 60, 'ISPHER': '1'}
[45]:
# The pyGamess supported basis options are listed by self.basis()
testDL.basis()
# Full configurations can be found in self.basisDict
basis = 'STO3G'
print(f'Settings for {basis}: {testDL.basisDict[basis]}')
PyGamess supported basis sets:
['STO3G', 'STO-3G', '321G', '3-21G', '631G', '6-31G', '6311G', '6-311G', '631G*', '6-31G*', '6-31G(D)', '631G(D)', '631G**', '6-31G**', '631GDP', '6-31G(D,P)', '631G(D,P)', '631+G**', '6-31+G**', '631+GDP', '6-31+G(D,P)', '631+G(D,P)', 'AM1', 'PM3', 'MNDO', 'note']
Settings for STO3G: {'gbasis': 'sto', 'ngauss': '3'}
[46]:
# Check updated input card
testDL.printGamessInput()
*** Gamess input card:
$contrl scftyp=rhf runtyp=energy maxit=60 ISPHER=1 $end
$basis gbasis=ACCD $end
$system mwords=30 $end
$DATA
N2O sym testing
CNV 8
O 8.0 0.0000000000 0.0000000000 1.2351270000
N 7.0 0.0000000000 0.0000000000 -1.1948180000
$END
[47]:
# Change some other parameters...
testDL.setParam(inputGroup='statpt', inputDict={'opttol': '0.01', 'nstep': '10'}) # Geom opt settings
testDL.params['system']['mwords']=50 # Memory settings, dict style
# Change atomList
# Note dictionary style will NOT work for 'extra' items, since some other settings are reconfigured in this case
testDL.setParam(inputGroup='extra', inputDict={'atomList':[0,1]})
Updating existing group 'statpt'. (To replace group, pass 'resetGroup=True')
Updated group 'statpt': {'opttol': '0.01', 'nstep': '10'}
Updating existing group 'extra'. (To replace group, pass 'resetGroup=True')
Updated group 'extra': {'job': 'N2O sym testing', 'sym': 'CNV 8', 'atomList': [0, 1]}
*** Found 'extra' job settings, updating local params and running setGamess()...
Set job from passed values.
Set sym from passed values.
Set atomList from passed values.
Set job = N2O sym testing
Set sym = CNV 8
Set atomList = [0, 1]
*** Gamess input card:
$contrl scftyp=rhf runtyp=energy maxit=60 ISPHER=1 $end
$basis gbasis=ACCD $end
$system mwords=50 $end
$DATA
N2O sym testing, ACCD
CNV 8
O 8.0 0.0000000000 0.0000000000 1.2351270000
N 7.0 0.0000000000 0.0000000000 -0.0403090000
$END
Additional transformations
Bond lengths & angles
These can be addressed using the RDkit functionality.
There is also a wrapper for bond lengths at the moment, self.setBondLength, which takes a dictionary of bonds to set, in the format {'Name':{'a1':0,'a2':1,'l':5}} where ‘a1’ and ‘a2’ give the atom indices for atoms defining the bond. This wraps RDkit’s rdkit.Chem.rdMolTransforms.SetBondLength.
TODO: clean up input, wrap angle setting functions(?).
Here’s a basic bond-length example
[48]:
testDL.printTable()
| Ind | Species | Atomic Num. | x | y | z | |
|---|---|---|---|---|---|---|
| 0 | 0 | O | 8 | 0.0 | 0.0 | 1.235127 |
| 1 | 1 | N | 7 | 0.0 | 0.0 | -0.040309 |
| 2 | 2 | N | 7 | 0.0 | 0.0 | -1.194818 |
[49]:
bonds = {'NO':{'a1':0, 'a2':1, 'l':5}, 'NN':{'a1':1, 'a2':2, 'l':2}}
# testDL.setBondLength(bonds = {'NO':{'a0':0, 'a1':1, 'l':5}})
testDL.setBondLength(bonds)
*** Set bonds, new coord table:
| Ind | Species | Atomic Num. | x | y | z | |
|---|---|---|---|---|---|---|
| 0 | 0 | O | 8 | 0.0 | 0.0 | 1.235127 |
| 1 | 1 | N | 7 | 0.0 | 0.0 | -3.764873 |
| 2 | 2 | N | 7 | 0.0 | 0.0 | -5.764873 |
Note that the RDkit routines move all atoms as appropriate for the new settings.
Manual coords
This is currently a little basic, and just wraps RDkit conformer.SetAtomPosition() routine for each atom. Set & select with a dictionary input, using atom indicies as keys.
[50]:
# Set coords for specified atoms
coordsRef = {0:[0,0,0], 1:[0.7,0,1.0]}
testDL.setCoords(coordsRef)
*** Updating coords (dictionary version).
*** Set atom positions, new coord table:
| Ind | Species | Atomic Num. | x | y | z | |
|---|---|---|---|---|---|---|
| 0 | 0 | O | 8 | 0.0 | 0.0 | 0.000000 |
| 1 | 1 | N | 7 | 0.7 | 0.0 | 1.000000 |
| 2 | 2 | N | 7 | 0.0 | 0.0 | -5.764873 |
[51]:
# Set all atoms on a specified axes with coord
# This can be useful for symmetrized cases, since otherwise atoms may be erroneously duplicated in Gamess run (even for near-zero coords)
testDL.setAxis({'x':0.0, 'y':0.0})
*** Set atom positions, new coord table:
| Ind | Species | Atomic Num. | x | y | z | |
|---|---|---|---|---|---|---|
| 0 | 0 | O | 8 | 0.0 | 0.0 | 0.000000 |
| 1 | 1 | N | 7 | 0.0 | 0.0 | 1.000000 |
| 2 | 2 | N | 7 | 0.0 | 0.0 | -5.764873 |
Minimal job pipeline to generate electronic structure & input files for ePolyScat
Possibly not advisable, since errors may go unnoticed, but the whole default pipeline can be executed with self.buildES(), which aims to string together the default cases above and produce a Gamess output file that can be used as input for ePolyScat.
This can be run directly at class creation by passing buildES=True. To keep the Gamess output file, pass fileOut.
Todo: fix issues with running for existing class object, may reset some items inconsistently at initGamess.
[52]:
# Set via build=True
# This runs the process from scratch
testBuild = ESgamess(searchName = 'N2O', fileOut = '/tmp/autobuild.out', buildES = True)
Set name = N2O
Set smiles = None
Set molFile = None
Set pd = None
Set xyz = None
Set molOverride = None
*** File /mnt/femtobackSSHFS/DriveSyncShare/code-share/github-share/epsman/docs/doc-source/demos/N2O.SDF already exists; existing file will be used. Pass overwrite=True to overwrite.
Set job = None
Set sym = C1
Set atomList = None
Set precision = 6
Set atomsDict = {}
Set refDict = {}
Set atomsHist = {}
*** Updating coords (Pandas version).
[14:15:24] Warning: molecule is tagged as 3D, but all Z coords are zero
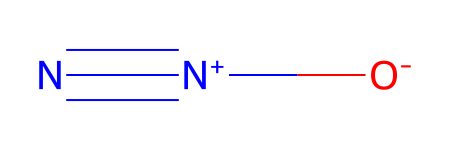
| Ind | Species | Atomic Num. | x | y | z | |
|---|---|---|---|---|---|---|
| 0 | 0 | O | 8 | 1.3063 | 0.0 | 0.0 |
| 1 | 1 | N | 7 | -0.1096 | 0.0 | 0.0 |
| 2 | 2 | N | 7 | -1.1967 | 0.0 | 0.0 |
*** Running default Gamess job.
*** Updating coords (Pandas version).
*** Set frame rotations, new coord table:
| Ind | Species | Atomic Num. | x | y | z | |
|---|---|---|---|---|---|---|
| 0 | 0 | O | 8 | 0.0 | 0.0 | 1.3063 |
| 1 | 1 | N | 7 | 0.0 | 0.0 | -0.1096 |
| 2 | 2 | N | 7 | 0.0 | 0.0 | -1.1967 |
Executeing py_rungms with command /opt/gamess/ddikick.x /opt/gamess/gamess.00.x pwqzab -ddi 1 1 jake -scr /tmp/tmp5d1rdu_t > /tmp/tmp5d1rdu_t/pwqzab.out
Executeing py_rungms with command /opt/gamess/ddikick.x /opt/gamess/gamess.00.x pwqzab -ddi 1 1 jake -scr /tmp/tmp5d1rdu_t > /tmp/tmp5d1rdu_t/pwqzab.out
INFO:pygamess.gamess:Executeing py_rungms with command /opt/gamess/ddikick.x /opt/gamess/gamess.00.x pwqzab -ddi 1 1 jake -scr /tmp/tmp5d1rdu_t > /tmp/tmp5d1rdu_t/pwqzab.out
*** Init pyGamess job.
Default Gamess input card set (use self.params to modify options dictionary, self.setGamess() to test):
$contrl scftyp=rhf runtyp=energy $end
$basis gbasis=sto ngauss=3 $end
$system mwords=30 $end
$DATA
None
C1
O 8.0 0.0000000000 0.0000000000 1.3063000000
N 7.0 0.0000000000 0.0000000000 -0.1096000000
N 7.0 0.0000000000 0.0000000000 -1.1967000000
$END
*** Found Gamess executable: /opt/gamess/gamess.00.x
*** ddikick exit status OK: ddikick.x: exited gracefully.
*** Gamess run completed OK.
E = -181.1830797993
*** Gamess output file moved to /tmp/autobuild.out
| Ind | Species | Atomic Num. | x | y | z | |
|---|---|---|---|---|---|---|
| 0 | 0 | O | 8 | 0.0 | 0.0 | 1.3063 |
| 1 | 1 | N | 7 | 0.0 | 0.0 | -0.1096 |
| 2 | 2 | N | 7 | 0.0 | 0.0 | -1.1967 |
[53]:
# Running the function for an existing class object also works
# Note this will use the existing geometry and configuration
# (although some items MAY BE reset by initGamess() routine - TBD)
testDL.buildES(fileOut = '/tmp/autobuild.out')
*** Running default Gamess job.
*** Updating coords (Pandas version).
*** Set frame rotations, new coord table:
| Ind | Species | Atomic Num. | x | y | z | |
|---|---|---|---|---|---|---|
| 0 | 0 | O | 8 | 0.0 | 0.0 | 1.588291 |
| 1 | 1 | N | 7 | 0.0 | 0.0 | 2.588291 |
| 2 | 2 | N | 7 | 0.0 | 0.0 | -4.176582 |
Executeing py_rungms with command /opt/gamess/ddikick.x /opt/gamess/gamess.00.x xnvffq -ddi 1 1 jake -scr /tmp/tmphj_nu26v > /tmp/tmphj_nu26v/xnvffq.out
Executeing py_rungms with command /opt/gamess/ddikick.x /opt/gamess/gamess.00.x xnvffq -ddi 1 1 jake -scr /tmp/tmphj_nu26v > /tmp/tmphj_nu26v/xnvffq.out
INFO:pygamess.gamess:Executeing py_rungms with command /opt/gamess/ddikick.x /opt/gamess/gamess.00.x xnvffq -ddi 1 1 jake -scr /tmp/tmphj_nu26v > /tmp/tmphj_nu26v/xnvffq.out
*** Init pyGamess job.
Default Gamess input card set (use self.params to modify options dictionary, self.setGamess() to test):
$contrl scftyp=rhf runtyp=energy $end
$basis gbasis=sto ngauss=3 $end
$system mwords=30 $end
$DATA
N2O sym testing
CNV 8
O 8.0 0.0000000000 0.0000000000 1.5882910000
N 7.0 0.0000000000 0.0000000000 2.5882910000
N 7.0 0.0000000000 0.0000000000 -4.1765820000
$END
*** Found Gamess executable: /opt/gamess/gamess.00.x
*** ddikick exit status OK: ddikick.x: exited gracefully.
*** Gamess run completed OK.
E = 0
*** Gamess output file moved to /tmp/autobuild.out
| Ind | Species | Atomic Num. | x | y | z | |
|---|---|---|---|---|---|---|
| 0 | 0 | O | 8 | 0.0 | 0.0 | 1.588291 |
| 1 | 1 | N | 7 | 0.0 | 0.0 | 2.588291 |
| 2 | 2 | N | 7 | 0.0 | 0.0 | -4.176582 |
Versions
[54]:
import scooby
scooby.Report(additional=['epsman', 'cclib', 'rdkit'])
[54]:
| Thu Jan 25 14:15:31 2024 EST | |||||
| OS | Linux | CPU(s) | 64 | Machine | x86_64 |
| Architecture | 64bit | Environment | Jupyter | ||
| Python 3.7.10 (default, Feb 26 2021, 18:47:35) [GCC 7.3.0] | |||||
| epsman | 0.0.1 | cclib | 1.7 | rdkit | 2023.03.2 |
| numpy | 1.19.2 | scipy | 1.6.1 | IPython | 7.21.0 |
| matplotlib | 3.3.4 | scooby | 0.5.6 | ||
| Intel(R) Math Kernel Library Version 2020.0.2 Product Build 20200624 for Intel(R) 64 architecture applications | |||||
[55]:
# Check current Git commit for local ePSproc version
from pathlib import Path
import epsman as em
!git -C {Path(em.__file__).parent} branch
!git -C {Path(em.__file__).parent} log --format="%H" -n 1
master
* restructure160221
bd21149f2aa6ee4ea23ab4dfd3f4f4a60503508e
[56]:
# Check current remote commits
!git ls-remote --heads https://github.com/phockett/epsman
21b4357a169baf9fa7887c68bd1cf8f92c59642c refs/heads/master
bd21149f2aa6ee4ea23ab4dfd3f4f4a60503508e refs/heads/restructure160221